Rare diseases
Hereditary Tyrosinemia Type-1
Tyrosinemia type 1 (HT1) is an inborn error of tyrosine catabolism caused by defective activity of fumarylacetoacetate hydrolase (FAH) and is characterized by progressive liver disease, renal tubular dysfunction, and porphyria-like symptoms.
History of the disease and its pharmacological treatment

The American biochemist Grace Medes (09/11/1886-31/12/1967) coins the term Tyrosinosis meanwhile describing the first biochemical findings in a 49-year-old man with myasthenia gravis. The paper entitled “A new error of tyrosine metabolism: Tyrosinosis” disclosed an unusual reducing substance in the urine of this patient which was identified as 4-hydroxyphenylpyruvate (4-HPP), assigning the defect in tyrosinosis to a deficiency of 4-hydroxyphenylpyruvate dioxygenase (4-HPPD). This condition was assigned the Mendelian Inheritance in Man code 76800.
Grace Medes wins the Garvan Medal, a prize that recognizes distinguished services to chemistry by women chemists.
Sakai and Kitagawa report the case of a two-year-old boy with marked hepatosplenomegaly having difficulties to thrive. Analysis of his urine revealed a large amount of 4-hydroxyphenyllactate (4-HPL) and a small amount of 4-HPP, 4-hydroxyphenylacetate (4-HPA) and tyrosine. The physical findings and biochemical observations led the authors to thinking that the patient presented a different inborn error of tyrosine metabolism from that reported a quarter of century before by Medes. They published their first paper on this case presenting it as “An atypical case of tyrosinosis”.
Third paper from Sakai, Kitagawa and Yoshioka, describing the pathological and biochemical observations of the organ tissues of this patient.
Prof. Sverre Halvorsen (24/07/1925-08/08/2012) and Leiv R. Gjessing disclose for the first time the upmost importance of a diet low in phenylalanine and tyrosine in the pathogenesis of the renal tubular lesions in a 2-year-old girl.
A symposium on tyrosinosis is organized in honour of Grace Medes by Prof. Sverre Halvorsen and Leiv R. Gjessing. It is of note that, Dr. Asbjorn Fölling, Professor Emeritus of the University of Oslo, who described the first case of phenylketonuria in 1934 attended the conference.
Dr. Jean Larochelle proposes to name the disease Hereditary Tyrosinemia.
Fumaryacetoacetate hydrolase (FAH) enzyme is identified as being the defective enzyme leading to HT-1. FAH is the last enzyme in the catabolic pathway of tyrosine.
Dr. Richard Gagné describes a method of detection of HT-1 as early as of 16 weeks of pregnancy by measuring the level of succinylacetone in the amniotic fluid.
The triketone herbicide Nitisinone, derived from leptospermone produced by the bottlebrush plant, is discovered by Zeneca Agrochemicals. Nitisinone is a 4-hydroxyphenylpyruvate dioxygenase (HPPD) inhibitor. HPPD is the second enzyme out of five, involved in the catabolic pathway of tyrosine, Fah being the last one. It is of note that, four of the five enzymes are associated with Inborn Errors of Metabolism (tyrosinemia type 2 and 3, hawkinsinuria, alkaptonuria and hyperphenylalaninemia due to dehydratase deficiency)
OMIM 276700 is assigned to tyrosinemia, type 1 (HT-1).
A critically ill 2-month-old baby becomes the first patient successfully treated with nitisinone for HT-1, after approval of a clinical trial in February by the Swedish Medical Agency. Then, more HT-1 patients were treated with nitisinone on a compassionate-use basis.

Sven Lindstedt (1927-2015) and Prof Elisabeth Holmes (1947-2015) describe that the use of nitisinone together with a proper diet correct and prevent neurological crisis deriving from HT-1. This discovery opened new hopes in patients who otherwise would have had to be transplanted.
Orfadin®, first treatment against HT-1 is approved by FDA in USA having nitisinone as active ingredient. Because of its instability at room temperature, Orfadin® has to be stored between 2-8°C.
Orfadin® is approved by EMA in Europe.
A working group from specialists in HT-1 chosen from Europe and Canada publishes the first recommendations for the management of HT-1.
The generic version Nitisinone Dipharma is approved in Europe, the first Nitisinone capsule formulation stable at room temperature.
Symptoms
Clinical symptoms typically begin before 2 years of age, with the majority of children presenting before the age of 6 months with evidence of acute liver failure and renal dysfunction. Neurologic crises, manifesting as painful episodes affecting extremities and/or abdominal function, accompanied by hypertension and hyponatremia, may present at any time and may result in respiratory failure and death. A few affected children may present over the age of 2 years with isolated coagulopathy or other signs of liver dysfunction, renal tubular disease, hypophosphatemic rickets, and failure to thrive. All children with HT-1 are at high risk for hepatocellular carcinoma.
Incidence
1 person in 100,000-120,000. This type of tyrosinemia is much more common in Quebec, Canada, and Scandinavia, where the overall incidence is about 1 in 20,000 individuals, with the Saguenay-Lac St. Jean region of Quebec, tyrosinemia type I even affects 1 in 1,846 people.
Treatment
If correctly identified and appropriately medically managed, the majority, if not all, of affected infants can anticipate a life free of hepatic and/or renal disease.
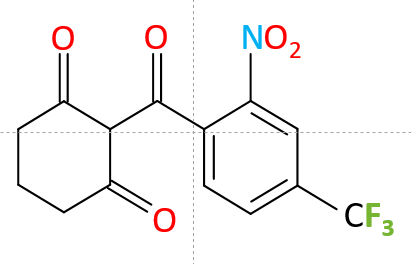
The primary treatment for type 1 tyrosinemia is nitisinone together with restriction of Tyrosine/Phenylalanine in the diet. Nitisinone, otherwise called 2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione (NTBC), is a potent inhibitor of 4-hydroxyphenylpyruvate dioxygenase (HPPD), which catalyses conversion of 4-hydroxyphenylpyruvate to homogentisic acid. By blocking the proximal tyrosinemia pathway, nitisinone minimizes the formation of fumarylacetoacetate and maleylacetoacetate. Since the availability of NTBC, liver transplantation as a treatment for HT-1 is limited to cases where the individual has a malignancy, and/or has decompensated liver disease refractory to NTBC.
Chinsky JM. Genet Med. 2017
Shaikh S et al. BJR Case Rep. 2018
 English
English Deutsch
Deutsch